minimod
Minimod is a simple tool for handling base modifications. It takes an aligned BAM file with modifications tags and the reference FASTA as inputs, and outputs base modifications (TSV) or base modification frequencies (TSV or bedmethyl).
Minimod reads base modification information encoded under MM:Z and ML:B:C SAM tags specified in SAMtags specification.
IMPORTANT: minimod is currently in active development. Open an issue if you find a problem or have a suggestion.


Table of Contents
- Quick start
- Installation
- Usage
- Examples
- minimod view
- minimod freq
- minimod summary
- Modification codes and contexts
- Modification threshold
- Enable insertions
- Enable haplotypes
- Important !
- Limitations / Future Improvements
- Acknowledgement
Quick start
If you are a Linux user and want to quickly try out download the compiled binaries from the latest release. For example:
VERSION=v0.4.0
wget "https://github.com/warp9seq/minimod/releases/download/$VERSION/minimod-$VERSION-x86_64-linux-binaries.tar.gz" && tar xvf minimod-$VERSION-x86_64-linux-binaries.tar.gz && cd minimod-$VERSION/
./minimod
Binaries should work on most Linux distributions as the only dependency is zlib which is available by default on most distributions. For compiled binaries to work, your operating system must have GLIBC 2.17 or higher (Linux distributions from 2014 onwards typically have this).
You can also use conda to install minimod as conda install minimod -c bioconda -c conda-forge.
Installation
Pre-requisites
sudo apt-get install zlib1g-dev # install zlib development libraries
Building a release
VERSION=v0.4.0
wget https://github.com/warp9seq/minimod/releases/download/$VERSION/minimod-$VERSION-release.tar.gz
tar xvf minimod-$VERSION-release.tar.gz
cd minimod-$VERSION/
scripts/install-hts.sh # download and compile the htslib
make
Usage
Usage information can be printed using minimod -h command.
Usage: minimod <command> [options]
command:
view view base modifications
freq output base modifications frequencies
summary output summary
Note: freq was previously mod-freq which still works but will be deprecated soon.
Examples
# view all 5mC methylations at CG context in tsv format (default mod code: m, context:CG)
minimod view ref.fa reads.bam > mods.tsv
# 5mC methylation frequencies at CG context in tsv format (default mod code: m, threshold: 0.8, context:CG)
minimod freq ref.fa reads.bam > modfreqs.tsv
# 5mC methylation frequencies at CG context in bedmethyl format (default mod code: m, threshold: 0.8, context:CG)
minimod freq -b ref.fa reads.bam > modfreqs.bedmethyl
# modification frequencies of multiple types ( m (5-methylcytosine) and h (5-hydroxymethylcytosine) in CG context with thresholds 0.8 and 0.7 respectively )
minimod freq -c m[CG],h[CG] -m 0.8,0.7 ref.fa reads.bam > mods.tsv
# summary of available modifications and counts
minimod summary reads.bam > summary.tsv
minimod view
minimod view ref.fa reads.bam > mods.tsv
This writes all base modifications (default modification code “m”) to a file (mods.tsv) in tsv format. Sample output is given below.
Usage: minimod view ref.fa reads.bam
basic options:
-c STR modification code(s) (eg. m, h or mh or as ChEBI) [m]
-t INT number of processing threads [8]
-K INT batch size (max number of reads loaded at once) [512]
-B FLOAT[K/M/G] max number of bases loaded at once [20.0M]
-h help
-p INT print progress every INT seconds (0: per batch) [0]
-o FILE output file [stdout]
--verbose INT verbosity level [4]
--version print version
--insertions enable modifications in insertions [no]
--haplotypes enable haplotype mode [no]
Sample mods.tsv output The output is ordered in the same as reads appear in the input BAM file, and for each read, entries are sorted by reference contig, reference position, strand, and modification code.
ref_contig ref_pos strand read_id read_pos mod_code mod_prob
chr22 19979864 + m84088_230609_030819_s1/55512555/ccs 14 m 0.709804
chr22 19979882 + m84088_230609_030819_s1/55512555/ccs 32 m 0.949020
chr22 19979885 + m84088_230609_030819_s1/55512555/ccs 35 m 0.980392
chr22 19979888 + m84088_230609_030819_s1/55512555/ccs 38 m 0.780392
chr22 19979900 + m84088_230609_030819_s1/55512555/ccs 50 m 0.623529
chr22 19979902 + m84088_230609_030819_s1/55512555/ccs 52 m 0.992157
chr22 19979929 + m84088_230609_030819_s1/55512555/ccs 79 m 0.941176
chr22 19979939 + m84088_230609_030819_s1/55512555/ccs 89 m 0.141176
chr22 19979948 + m84088_230609_030819_s1/55512555/ccs 98 m 0.623529
| Field | Type | Definition |
|---|---|---|
| 1. ref_contig | str | chromosome |
| 2. ref_pos | int | position (0-based) of the base in reference |
| 3. strand | char | strand (+/-) of the read |
| 4. read_id | str | name of the read |
| 5. read_pos | int | position (0-based) of the base in read |
| 6. mod_code | char | base modification code as in SAMtags: 1.7 Base modifications |
| 7. mod_prob | float | probability (0.0-1.0) of base modification |
| 8. ins_offset | int | offset of inserted base from ref_pos (only output when –insertions is specified) |
| 9. haplotype | int | haplotype of the read (only output when –haplotypes is specified) |
minimod freq
minimod freq ref.fa reads.bam > modfreqs.tsv
This writes base modification frequencies (default modification code “m” in CG context with modification threshold 0.8) to a file (modfreqs.tsv) file in tsv format.
Usage: minimod freq ref.fa reads.bam
basic options:
-b output in bedMethyl format [not set]
-c STR modification code(s) (eg. m, h or mh or as ChEBI) [m]
-m FLOAT min modification threshold(s). Comma separated values for each modification code given in -c [0.8]
-t INT number of processing threads [8]
-K INT batch size (max number of reads loaded at once) [512]
-B FLOAT[K/M/G] max number of bases loaded at once [20.0M]
-h help
-p INT print progress every INT seconds (0: per batch) [0]
-o FILE output file [stdout]
--verbose INT verbosity level [4]
--version print version
--insertions enable modifications in insertions [no]
--haplotypes enable haplotype mode [no]
Sample modfreqs.tsv output The output entries are sorted by reference contig, reference position, strand, and modification code.
contig start end strand n_called n_mod freq mod_code
chr22 20016337 20016337 + 5 0 0.000000 m
chr22 20016594 20016594 + 2 0 0.000000 m
chr22 20017045 20017045 + 1 0 0.000000 m
chr22 19970705 19970705 + 1 0 0.000000 m
chr22 19981716 19981716 + 1 1 1.000000 m
chr22 20020909 20020909 + 3 0 0.000000 m
chr22 19995719 19995719 + 4 2 0.500000 m
chr22 20017060 20017060 + 1 0 0.000000 m
chr22 19971259 19971259 + 1 1 1.000000 m
| Field | Type | Definition |
|---|---|---|
| 1. contig | str | chromosome |
| 2. start | int | position (0-based, inclusive) of the base |
| 3. end | int | position (0-based, inclusive) of the base |
| 4. strand | char | strand (+/-) of the read |
| 5. n_called | int | number of reads called for base modification |
| 6. n_mod | int | number of reads with base modification |
| 7. freq | float | n_mod/n_called ratio |
| 8. mod_code | char | base modification code as in SAMtags: 1.7 Base modifications |
| 9. ins_offset | int | offset of inserted base from ref_pos (only output when –insertions is specified) |
| 10. haplotype | int | haplotype of the read (only output when –haplotypes is specified) |
Sample modfreqs.bedmethyl output
chr22 20016387 20016388 m 4 - 20016387 20016388 255,0,0 4 0.000000
chr22 20016820 20016821 m 1 + 20016820 20016821 255,0,0 1 0.000000
chr22 19999255 19999256 m 7 + 19999255 19999256 255,0,0 7 0.000000
chr22 20016426 20016427 m 1 + 20016426 20016427 255,0,0 1 100.000000
chr22 19988365 19988366 m 1 - 19988365 19988366 255,0,0 1 100.000000
chr22 19988168 19988169 m 1 - 19988168 19988169 255,0,0 1 100.000000
chr22 20016904 20016905 m 1 + 20016904 20016905 255,0,0 1 0.000000
chr22 20011898 20011899 m 8 - 20011898 20011899 255,0,0 8 25.000000
chr22 19990123 19990124 m 3 + 19990123 19990124 255,0,0 3 0.000000
chr22 19982787 19982788 m 1 + 19982787 19982788 255,0,0 1 0.000000
| Field | Type | Definition |
|---|---|---|
| 1. contig | str | chromosome |
| 2. start | int | position (0-based, inclusive) of the base |
| 3. end | int | position (0-based, not inclusive) of the base |
| 4. mod_code | char | base modification code as in SAMtags: 1.7 Base modifications |
| 5. n_mod | int | number of reads with base modification |
| 6. strand | char | strand (+/-) of the read |
| 7. start | int | = field 2 |
| 8. end | int | = field 3 |
| 9. n_mod | int | = field 5 |
| 10. freq | float | n_mod/n_called ratio |
minimod summary
minimod summary reads.bam > summary.tsv
This writes all base modifications available to a file (summary.tsv) in tsv format. Sample output is given below.
Usage: minimod summary reads.bam
basic options:
-t INT number of processing threads [8]
-K INT batch size (max number of reads loaded at once) [512]
-B FLOAT[K/M/G] max number of bases loaded at once [20.0M]
-h help
-p INT print progress every INT seconds (0: per batch) [0]
-o FILE output file [stdout]
--verbose INT verbosity level [4]
--version print version
advanced options:
--debug-break INT break after processing the specified no. of batches
--profile-cpu=yes|no process section by section
Sample mods.tsv output
read_id modifications
491fb526-314e-4c18-9690-eb6930d780ea A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
52b66d0d-a21e-4334-be1b-f72486d9f9bf A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
cb481f14-7651-448c-945b-b4f5b2e8b70c A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
2cab7053-9008-47eb-8e57-c33fda56c2ec A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
37bf1305-8d8b-4973-9a0b-930303067306 A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
164e336f-568d-44d5-882d-7669bbe67654 A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
229bbbb9-abf9-4825-af30-a583a19864eb A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
adbaba61-604d-4897-99cc-f9a934f3e2c8 A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
093bc2c6-2ae5-437d-a17c-be2755c3c689 A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
92fc0cc5-b2d8-4cb1-bf25-8f8b88088f23 A|17596|. C|m|. A|a|. C|19228|. T|19227|. A|69426|. G|19229|. T|17802|.
The output is ordered in the same as reads appear in the input BAM file. First column of the output are the read_id the second column contains all available modifications in that read as space separated entries. Each entry has the following format.
canonical_base(character such as ACGTN)|mod_code(character or ChEBI number)|status_flag(. or ?)
Status flag describes how skipped bases (not included in the output of view or freq subtools) should be interpreted by downstream tools.
- . : skipped bases should be assumed to have low probability of modifications.
- ? : there is no information about the modification status of skipped bases
Modification codes and contexts
Base modification codes and contexts can be set for both view and freq tool using -c option to take only specific base modifications found in a given contexts. The context should match in the reference and bases in unmatching contexts are ignored.
Here are the possible context formats.
- a[A] : type a modifications of all A bases
- h[CG] : type h modifications in CG context (CpG sites)
- m : type m modifications in default CG context
- a[*] : type a modifications in all contexts
- *[CG] : all types of modifications in CG context
- * : all types of modifications in all contexts
- 17802[T] : pseU modifications in T context (modification code is given as ChEBI code)
Here are some example commands.
minimod view -c a[A],h[CG],m,a[*] ref.fa reads.bam
minimod freq -c a[A],h[CG],m,a[*] ref.fa reads.bam
minimod freq -c "*[CG]" ref.fa reads.bam
minimod freq -c "*" ref.fa reads.bam
minimof freq -c "17802[T]" ref.fa reads.bam
If the context is not specified in square brackets along with modification code, minimod will consider following default contexts.
All possible modification codes are supported by minimod along with default contexts if not specified (SAMtags: 1.7 Base modifications)
| Unmodified base | Code | Abbreviation | Name | Default context |
|---|---|---|---|---|
| C | m | 5mC | 5-Methylcytosine | CG |
| C | h | 5hmC | 5-Hydroxymethylcytosine | CG |
| A | a | 6mA | 6-Methyladenine | A |
| U | 17802 | pseU | Pseudouridine | U |
Note that we have done a lot of testing on 5mc and some limited testing on 6mA and 5hmC. The others are not yet tested.
Modification threshold
Base modification threshold can be set for freq tool using -m option.
- 5mC modification(default context :CG) frequencies with threshold 0.8
minimod freq -c m -m 0.8 ref.fa reads.bam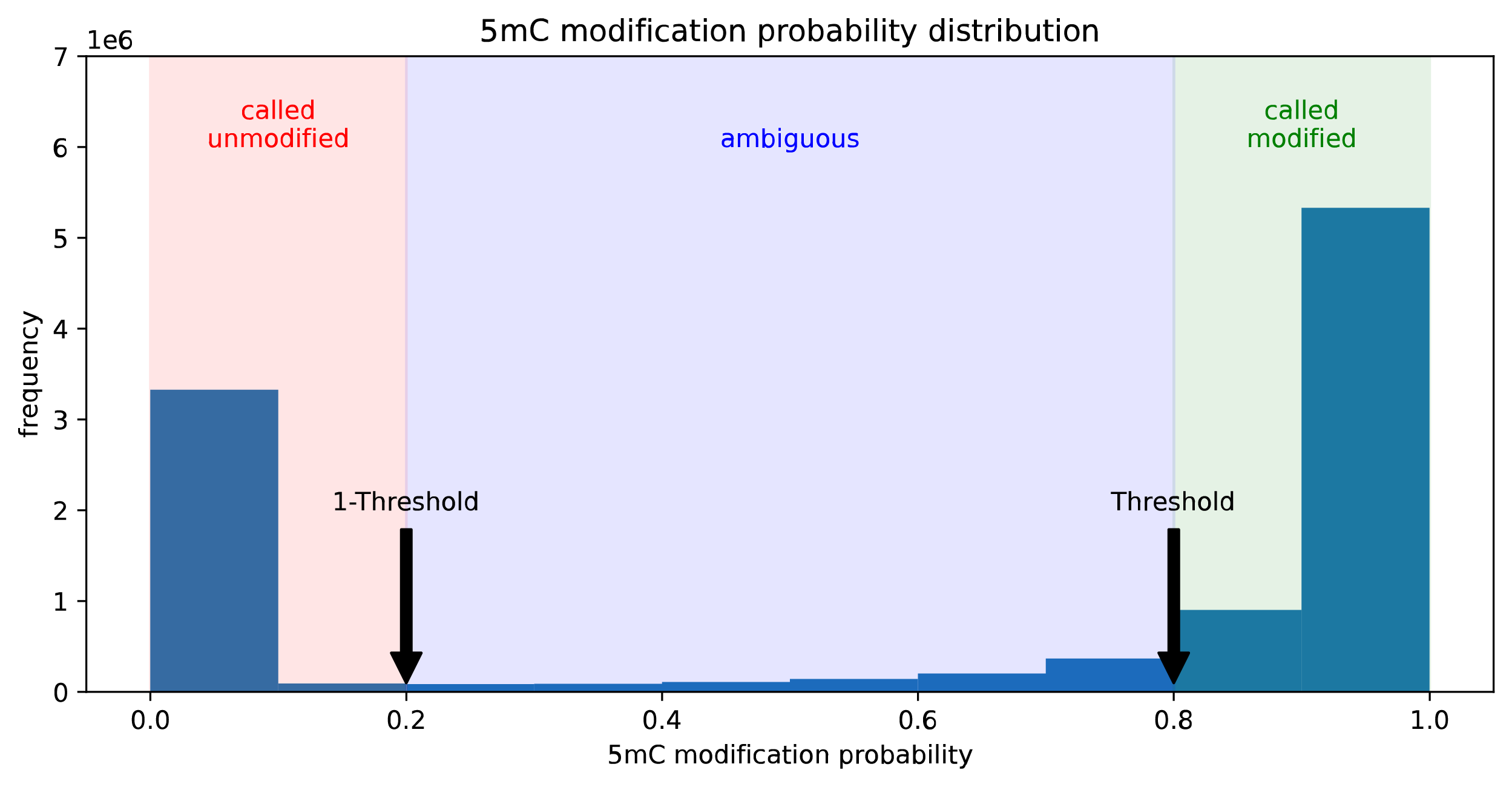
If p(5mC) >= 0.8 (threshold), called(5mC) and modified(5mC) If p(5mC) <= 0.2 (1-threshold), called(5mC) else, ignored as ambiguous freq(5mC) = total_modified(5mC)/total_called(5mC) - 5mC and 5hmC base modification(default context :CG) frequencies with thresholds 0.8, 0.7 respectively
minimod freq -c m,h -m 0.8,0.7 ref.fa reads.bam
If p(5mC) >= 0.8 (threshold), called(5mC) and modified(5mC) If p(5mC) <= 0.2 (1-threshold), called(5mC) else, ambiguous freq(5mC) = total_modified(5mC)/total_called(5mC)
If p(5hmC) >= 0.7 (threshold), called(5hmC) and modified(5hmC) If p(5hmC) <= 0.3 (1-threshold), called(5hmC) else, ignored as ambiguous freq(5hmC) = total_modified(5hmC)/total_called(5hmC)
Enable insertions
minimod can handle inserted modified bases (where canonical base in not in reference) by specifying –insertions flag for both freq and view tools.
Specifying –insertions will add an extra ins_offset column(only in tsv output) which is the position of modified base within the inserted region.
Sample output of view with –insertions
$ minimod view --insertions ref.fa reads.bam
ref_contig ref_pos strand read_id read_pos mod_code mod_prob ins_offset
chr22 19967897 + 89870c83-8790-419f-acf8-8a8e93a0f3c9 1396 m 0.537255 0
# chr22 19968083 + 89870c83-8790-419f-acf8-8a8e93a0f3c9 1582 m 0.000000 2
chr22 19968225 + 89870c83-8790-419f-acf8-8a8e93a0f3c9 1722 m 0.105882 0
chr22 19968330 + 89870c83-8790-419f-acf8-8a8e93a0f3c9 1826 m 0.505882 0
chr22 19968390 + 89870c83-8790-419f-acf8-8a8e93a0f3c9 1885 m 0.960784 0
chr22 19968435 + 89870c83-8790-419f-acf8-8a8e93a0f3c9 1930 m 0.917647 0
Sample output of freq with –insertions
$ minimod freq --insertions ref.fa reads.bam
contig start end strand n_called n_mod freq mod_code ins_offset
chr22 19981825 19981825 - 1 1 1.000000 m 0
# chr22 19968083 19968083 + 1 0 0.000000 m 2
chr22 20014485 20014485 + 1 1 1.000000 m 0
chr22 20017214 20017214 - 1 0 0.000000 m 0
chr22 20004425 20004425 + 2 2 1.000000 m 0
chr22 20016700 20016700 - 4 0 0.000000 m 0
Highlighted line corresponds to a 5mC modification within an insertion (A mC G) at position 19968083
Enable haplotypes
minimod can output the haplotype in a separate integer column (only in tsv output) by specifying –haplotypes flag for both view and freq tools. minimod does not compute or infer haplotypes. Instead, it uses haplotype assignments already present in the input BAM, if the BAM is phased and contains the HP (Haplotype) tag.
Sample output of view with –haplotypes
$ minimod view --haplotypes ref.fa reads.bam
ref_contig ref_pos strand read_id read_pos mod_code mod_prob haplotype
chr1 10484 - m84088_240522_013656_s1/164692234/ccs 16262 m 0.980392 2
chr1 10471 - m84088_240522_013656_s1/164692234/ccs 16275 m 0.772549 2
chr1 10469 - m84088_240522_013656_s1/164692234/ccs 16277 m 0.254902 2
chr1 25115 - m84088_240522_013656_s1/197203096/ccs 0 m 0.290196 1
chr1 25059 - m84088_240522_013656_s1/197203096/ccs 56 m 0.886275 1
chr1 24926 - m84088_240522_013656_s1/197203096/ccs 189 m 0.988235 1
Sample output of freq with –haplotypes
$ minimod freq --haplotypes ref.fa reads.bam
contig start end strand n_called n_mod freq mod_code haplotype
chr1 23002 23002 - 3 3 1.000000 m 1
chr1 23002 23002 - 3 3 1.000000 m 2
chr1 23002 23002 - 6 6 1.000000 m *
chr1 23096 23096 + 1 0 0.000000 m 1
chr1 23096 23096 + 3 3 1.000000 m 2
chr1 23096 23096 + 4 3 0.750000 m *
freq value of modifications with haplotype=* is calculated taking modifications from all haplotypes
Important !
Make sure that you handle the modification tags correctly in each step in base modification calling pipeline (e.g., providing both -y and -Y to minimap2). See the example pipeline that we use below.
Base-calling
-
Use a basecalling model trained to identify modified bases.
Example: Base-calling a slow5 file using buttery-eel
buttery-eel --call_mods --config dna_r10.4.1_e8.2_400bps_5khz_modbases_5hmc_5mc_cg_hac.cfg -i reads.slow5 -o reads.sam -g path/to/guppy/bin samtools fastq -TMM,ML reads.sam > reads.fastq
Aligning
- Avoid secondary alignments
-
Use soft clipping for supplementary alignments
Corresponding minimap2 flags are as follows. | Minimap2 Flag | Description | |-|-| |-Y | Use soft clipping for supplementary alignments | |-y | Copy input FASTA/Q comments to output | |–secondary=no| Avoid secondary alignments |
Example: aligning ONT reads using minimap2
minimap2 -ax map-ont -Y -y --secondary=no ref.idx reads.fastq - If more than 90% of the reads in the BAM file are skipped due to various reasons (unmapped, 0 length, or missing MM/ML tags), minimod prints a warning message. However, if all of them are skipped minimod errors out.
- If hard clipped non-primary alignments are found, minimod errors out. (to filter out non-primary alignments:
samtools view -h -F 2308 reads.bam -o primary_reads.bamor use minimap2 with -Y to use soft clipping).
Limitations / Future Improvements
- Status of skipped bases (encoded as . or ? in MM tag) are ignored
Acknowledgement
Minimod uses klib. Some code snippets have been taken from Minimap2 and Samtools.
Citation
Samarasinghe, S., Deveson, I., Gamaarachchi, H., 2025. Base modification analysis in long read sequencing data using Minimod. https://doi.org/10.1101/2025.07.16.665072
@article{samarasinghe_base_2025,
title = {Base modification analysis in long read sequencing data using {Minimod}},
author = {Samarasinghe, Suneth and Deveson, Ira and Gamaarachchi, Hasindu},
year={2025},
publisher={bioRxiv}
}